Ambas as drogas já foram aprovadas nas fases 1 e 2 dos testes clínicos e demonstraram serem seguras para o consumo humano.
Com testes em laboratório feitos em minicérebros humanos, uma equipe de pesquisadores liderados pelo neurocientista brasileiro Alysson Muotri, na Universidade da Califórnia em San Diego (EUA), conseguiu reverter várias características da Síndrome de Rett e já conta com dois medicamentos para iniciar testes clínicos na fase três (já aprovados nas fases 1 e 2, demonstrando serem seguros para o consumo humano). Os minicérebros “tratados” passaram a se comportar como se não tivessem a Síndrome de Rett.
A maioria das condições de saúde ligadas ao Transtorno do Espectro do Autismo tem um componente genético complexo e multifatorial. A Síndrome de Rett é uma exceção. Os bebês nascidos com essa forma do transtorno apresentam mutações específicas no gene MECP2, causando um prejuízo grave no desenvolvimento do cérebro que afeta principalmente as mulheres e, geralmente, vem acompanhado de características autísticas. No entanto, ainda não há tratamento para a causa — as terapias atuais visam aliviar os sintomas e obter ganho em qualidade de vida. Muitos indivíduos com Rett estão dentro do espectro do autismo.
O que os pesquisadores do Muotri Lab — da Universidade da Califórnia em San Diego (UCSD) e do Consórcio Sanford para Medicina Regenerativa — fizeram recentemente foi usar “minicérebros” (tecnicamente organoides cerebrais derivados de células-tronco) com mutação no gene MECP2 para melhor estudar a síndrome.
Um estudo tão detalhado e tão próximo de um cérebro real só foi possível porque a equipe de Muotri conseguiu, recentemente, otimizar a tecnologia de construção de organoides do cérebro para corresponder ao padrão de impulso elétrico de bebês prematuros (fazendo, inclusive, exames de eletroencefalograma nos minicérebros e registrando atividades cerebrais), tornando-os mais parecidos com cérebros humanos reais do que nunca.
A startup brasileira de biotecnologia Tismoo, da qual Muotri é um dos cofundadores, tem planos de implantar sua unidade de testes de medicamentos (drug discovery), utilizando essa mesma plataforma tecnológica de minicérebros, a partir de 2023.
 Pesquisa publicada
Pesquisa publicada
Em um estudo publicado dia 8 de dezembro de 2020, na revista científica EMBO Molecular Medicine, a equipe identificou dois medicamentos candidatos a neutralizar os déficits causados pela falta do gene MECP2. Esses compostos restauraram os níveis de cálcio, a produção de neurotransmissores e a atividade do impulso elétrico, fazendo os minicérebros com Rett se comportar como se não tivessem a síndrome, segundo Muotri.
“A mutação do gene que causa a Síndrome de Rett foi descoberta décadas atrás, mas o progresso no seu tratamento tem demorado. Pelo menos em parte, isso aconteceu porque os estudos com modelos de camundongos não obtiveram os mesmos resultados para os humanos”, disse o líder do estudo, o neurocientista brasileiro Alysson Muotri, professor de pediatria e medicina celular e molecular na faculdade de medicina da Universidade da Califórnia em San Diego (EUA). “Este estudo foi impulsionado pela necessidade de um modelo que imitasse melhor o cérebro humano”, explicou.
Organoides cerebrais, os minicérebros, são modelos celulares tridimensionais que representam aspectos do cérebro humano em laboratório. Esses organoides ajudam os pesquisadores a rastrear o desenvolvimento humano, desvendar os eventos moleculares que levam a doenças e transtornos, além de testar novos tratamentos. Na UCSD, os organoides cerebrais foram usados para produzir a primeira prova experimental direta de que o vírus Zika brasileiro pode causar defeitos congênitos graves e para recolocar os medicamentos existentes para o HIV no tratamento de um outro distúrbio neurológico hereditário raro. Muotri e sua equipe também enviaram minicérebros para a Estação Espacial Internacional a fim de testar o efeito da microgravidade no desenvolvimento do cérebro — e as perspectivas de vida humana fora da Terra.
Eles não são réplicas perfeitas do cérebro humano, é claro. Os organoides não têm conexões com outros sistemas orgânicos, como vasos sanguíneos. Drogas testadas em organoides cerebrais são adicionadas diretamente neles — as substâncias não precisam atravessar a barreira hematoencefálica, vasos sanguíneos especializados em manter o cérebro praticamente livre de bactérias, vírus e toxinas.
Muito próximo do real
Mas os pesquisadores consideram os organoides muito úteis para verificar mudanças na estrutura física ou na expressão do gene ao longo do tempo, ou ainda o efeito de uma mutação genética, vírus ou droga.
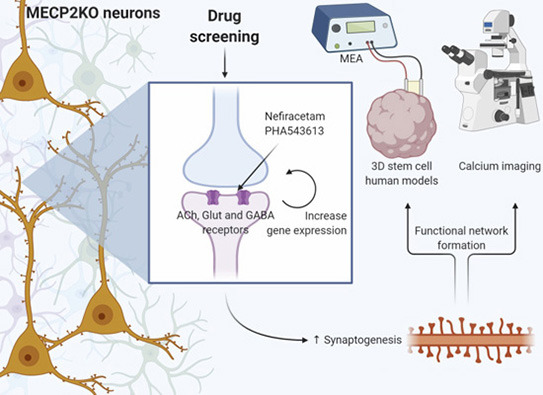
No último estudo, os pesquisadores aplicaram este novo protocolo para organoides cerebrais funcionais, usando células-tronco pluripotentes induzidas (iPSCs, na sigla em inglês) derivadas de pacientes com Síndrome de Rett. Em suma, eles coletaram uma amostra de pele, trataram as células com uma técnica que as converteu em iPSCs. A partir daí, como passam a ser células-tronco, podendo se transformar em qualquer célula do corpo, elas são induzidas a se tornarem células cerebrais (neurônios), preservando a origem genética única de cada paciente. Para verificar suas descobertas, a equipe também desenvolveu organoides cerebrais artificialmente sem o gene MECP2, e até mesmo misturou células mutadas e de controle (de neurotípicos) para simular o padrão de mosaico normalmente visto em pacientes do sexo feminino.
A falta do gene MECP2 mudou muita coisa nos minicérebros: forma, subtipos de neurônios presentes, padrões de expressão gênica, produção de neurotransmissores e formação de sinapses. A atividade do cálcio e os impulsos elétricos também diminuíram. Essas mudanças levaram a grandes defeitos no surgimento de ondas oscilatórias neurais corticais, também conhecidas como “ondas cerebrais”.
Em uma tentativa de compensar a falta do gene MECP2, a equipe tratou os organoides do cérebro com 14 drogas candidatas que são conhecidas por afetar várias funções das células cerebrais. Quase todos os sintomas moleculares e celulares foram resolvidos quando os pesquisadores trataram os organoides do cérebro da Síndrome de Rett com as duas melhores drogas candidatas: Nefiracetam e PHA 543613. O número de neurônios ativos nos organoides da Síndrome de Rett, por exemplo, praticamente dobrou após o tratamento. O Nefiracetam e o PHA 543613 foram testados anteriormente em ensaios clínicos de fase 1 e 2 para o tratamento de outras doenças, o que significa que já se sabe que atravessam a barreira hematoencefálica e são seguros para consumo humano.
De acordo com Muotri, esses resultados laboratoriais fornecem um argumento convincente para o avanço do Nefiracetam e do PHA 543613 em ensaios clínicos para pacientes com distúrbios do neurodesenvolvimento relacionados a mutações no gene MECP2.
Coquetéis de drogas
No final, o melhor tratamento para a Síndrome de Rett pode não ser uma “super” droga, disse Muotri, que também é diretor do Programa de Células-Tronco da UCSD, membro do o Consórcio Sanford para Medicina Regenerativa, cofundador da Tismoo e da rede social Tismoo.me. “Há uma tendência no campo da neurociência de procurar medicamentos altamente específicos que atinjam alvos exatos, e de usar um único medicamento para uma doença complexa”, explicou ele.. “Mas não fazemos isso para muitos outros distúrbios complexos, onde tratamentos multifacetados são usados. Da mesma forma, aqui nenhum alvo resolveu todos os problemas. Precisamos começar a pensar em termos de coquetéis de drogas, assim como têm sido bem-sucedidos no tratamento de HIV e câncer”, argumentou.
Também são coautores do estudo: Cleber A. Trujillo, Jason W. Adams, Leon Tejwani, Allan Acab, Charles A. Thomas, UC San Diego; Priscilla D. Negraes, Cassiano Carromeu, UC San Diego e StemoniX Inc.; Ben Tsuda, Terrence J. Sejnowski, UC San Diego e Salk Institute for Biological Studies; Neha Sodhi, Katherine M. Fichter, Fabian Zanella, StemoniX Inc.; Henning Ulrich, Universidade de São Paulo.
Estudo completo
O estudo na íntegra pode ser obtido em https://www.embopress.org/doi/full/10.15252/emmm.202012523 ou leia a versão em PDF.
Lei também nosso artigo “Muotri envia 2ª etapa de sua pesquisa com minicérebros humanos para o espaço“.
 Francisco Paiva Jr. / Revista Autismo
Francisco Paiva Jr. / Revista Autismo
 Tismoo.me
Tismoo.me Aplicativo
Aplicativo